Detection of Copy Number Variation in Targeted Sequencing Samples
Hong Zheng / 2017-08-28
Introduction
There are two categories of methods for copy number variation (CNV) detection.
Based on read pair information
1) The paired-end mapping approach, in which mapped paired-reads whose distances are significantly different from the predetermined average insert size are used.
Tools: BreakDancer, VariationHunter, PEMer, etc.
2) The split read (SR) approach identifies read pairs in which one read from each pair is aligned to the reference genome uniquely while the other one fails to map or only partially maps to the genome. Those unmapped or partially mapped reads potentially provide accurate breaking points at the single base pair level for CNVs or structural variants.
Tools: Pindel, BreakSeq2, DellyBased on read depth (RD) information
With the accumulation of high-coverage sequencing data, RD-based methods have become a major approach to estimate CNV. The underlying hypothesis is that the read depth of a genomic region is correlated with the copy number of the region.
1) For whole-genome sequencing (WGS), a sliding window approach is adopted.
Tools: CNVnator, CNVnorm, cn.MOPS, cnvHMM, CNV-seq, JointSLMb, etc.
2) For whole-exome sequencing (WES), due to differing capture efficiency and non-continuous distributions of the reads, the methods used for WGS are not suitable. Several special methods have been developed for WES.
a) multiple samples as input. XHMM, CODEX, CoNIFER, ExoCNVTest
b) case-control samples as input. ExomeCNV, CONTRA, PropSeq, VarScan2
c) Others. ExomeDepth, CONDEX, SeqGene, Control-FREEC
The read pair-based methods rely heavily on the location of breakpoints, CNV size relative to insert size, and read length. Due to the discontinuation of genomic regions in exome/targeted sequencing, most CNV breakpoints could not be detected using these methods.
In contrast, RD-based methods are good for detection of CNVs in scarce genomic regions. Moreover, RD-based methods can detect large insertions and CNVs in complex genomic regions, which are difficult to detect using read pair-based methods.
For RD-based methods, instead of looking at a single sample one time, more and more tools utilize multiple samples to increase sensitivity and reduce false discovery.
In this demo, both the read pair-based and RD-based methods were deployed to investigate the CNVs in targeted sequencing data. The methods that have been used are emphasized in bold font in the text.
Results
Ground-truth 1000 Genome CNV data
########
#download
########
wget ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Homo_sapiens/by_study/estd219_1000_Genomes_Consortium_Phase_3_Integrated_SV/vcf/estd219_1000_Genomes_Consortium_Phase_3_Integrated_SV.GRCh37.submitted.variant_call.germline.vcf.gz
########
#process
########
pre=estd219_1000_Genomes_Consortium_Phase_3_Integrated_SV.GRCh37.submitted.variant_call.germline
zcat $pre.vcf.gz | egrep -v '^#' | sed 's/;/\t/g' | grep -v CIPOS | awk 'OFS="\t"{print $1,$2,$13,$12,$10,$9}' | sed 's/END=//;s/SAMPLE=//;s/SVTYPE=//;s/CALLID=//' > $pre.preciseCNV.txt
zcat $pre.vcf.gz | egrep -v '^#' | sed 's/;/\t/g' | grep CIPOS | awk 'OFS="\t"{print $1,$2,$15,$12,$10,$9}' | sed 's/END=//;s/SAMPLE=//;s/SVTYPE=//;s/CALLID=//' > $pre.impreciseCNV.txt
cat $pre.preciseCNV.txt $pre.impreciseCNV.txt > $pre.CNV.txt
XHMM
XHMM takes multiple samples as input. The demo is based on targeted sequencing data of 105 genes in 118 samples.
1) Calculate depth of coverage from BAM files
Input:
- A list of BAM files, generated properly from the established pipeline for variant calling.
- The BED file of targeted regions in proper format. The chromosome names should match with the BAM files. The chromosome names and starting coordinates should be sorted. Redundant or overlapping regions should be merged (bedtools merge). The choice of regions in the BED file is fairly important, as it is the basic unit for coverage calculation and CNV calling.
Scripts:
run_DepthOfCoverage.sh:
work_dir=/home/zhengh/projects/CNV/105genes
target=105genespure
interval_file=/home/database/targetBed/$target.bed
/home/pipelines/DepthOfCoverage.sh \
$work_dir/bam.list $interval_file $work_dir/QC $target \
1> $work_dir/logs/$target.DepthOfCoverage.log 2>&1
DepthOfCoverage.sh:
bamlist=$1
interval_file=$2
QC_dir=$3
interval=$4
DIR="${BASH_SOURCE%/*}";if [[ ! -d "$DIR" ]]; then DIR="$PWD"; fi
source "$DIR/DATABASESandTOOLS.sh"
###-------Tutorial------------------------------------------------------------------------------------------------------
### http://gatkforums.broadinstitute.org/gatk/discussion/40/performing-sequence-coverage-analysis
###-------Tutorial------------------------------------------------------------------------------------------------------
###-------DepthOfCoverage-----------------------------------------------------------------------------------------------
echo "Goodluck! DepthOfCoverage started on `date`"
$java -Xmx8G -jar $gatk -T DepthOfCoverage \
-I $bamlist \
-R $ref_dir/human_g1k_v37_decoy.fasta \
-L $interval_file \
-o $QC_dir/$interval.bam.DP \
--minBaseQuality 0 --minMappingQuality 20 \
--start 1 --stop 5000 --nBins 200 \
-dt BY_SAMPLE -dcov 5000 \
--includeRefNSites --countType COUNT_FRAGMENTS \
-geneList $ref_dir/hg19.geneTrack.refSeq
echo "Goodluck! DepthOfCoverage finished on `date`"
###-------DepthOfCoverage-----------------------------------------------------------------------------------------------
Output:
Target total_coverage average_coverage HG00707_S2_total_cvg HG00707_S2_mean_cvg HG00707_S2_granular_Q1 HG00707_S2_granular_median HG00707_S2_granular_Q3 HG00707_S2_%_above_15
1:17345334-17345466 662773 4983.26 12917 97.12 94 99 104 100.0
1:17349079-17349268 878128 4621.73 16233 85.44 78 87 97 100.0
1:17350434-17350584 499734 3309.50 9242 61.21 58 64 69 100.0
1:17354219-17354360 669584 4715.38 13648 96.11 90 96 105 100.0
1:17355074-17355252 1200155 6704.78 22278 124.46 109 129 146 100.0
1:17359524-17359658 849562 6293.05 15855 117.44 108 123 134 100.0
2) XHMM calling
2.1) Change data format, remove samples and target regions that do not fulfill coverage requirments, and mean-center the coveage.
a) The”chr” are added to chromosome names to facilitate plots.
b) Here the default thresholds for coverage are used.
target mean coverage: between 10 and 500
sample mean coverage: between 25 and 200
In this case, 17 samples and 15 targets are removed due to low coverage. They are found in files ending with “filtered_centered.RD.txt.filtered_targets.txt” and “filtered_centered.RD.txt.filtered_samples.txt”.
Importantly, if the experimental setup is expected to result in mean depths that deviate from these generic values, then these values need to be changed to reflect the dataset and only remove true outlier targets and samples. The default values work well for the current dataset.
c) Center each column of the matrix (RD.txt) so that the mean target value is 0 in preparation for PCA based normalization in the next step.
work_dir=/home/zhengh/projects/CNV/105genes
xhmm=/home/tools/XHMM/statgen-xhmm-cc14e528d909/xhmm
covdata=$work_dir/QC/105genespure.bam.DP.sample_interval_summary
pre=118bam.105genespure
ref=/home/database/b37/human_g1k_v37_decoy.fasta
target=105genespure
interval_file=/home/database/targetBed/$target.bed
param=/home/tools/XHMM/statgen-xhmm-cc14e528d909/params.txt
param_adjusted=/home/zhengh/projects/CNV/105genes/xhmm/params.txt
########
echo "change data format. Good luck."
########
less $covdata | sed '2,$s/^/chr/' > $work_dir/xhmm/$pre.DP.sample_interval_summary
$xhmm --mergeGATKdepths -o $work_dir/xhmm/$pre.RD.txt --GATKdepths $work_dir/xhmm/$pre.DP.sample_interval_summary
#######
echo "mean-centers the targets. Good luck."
#######
$xhmm --matrix -r $work_dir/xhmm/$pre.RD.txt --centerData --centerType target \
-o $work_dir/xhmm/$pre.filtered_centered.RD.txt \
--outputExcludedTargets $work_dir/xhmm/$pre.filtered_centered.RD.txt.filtered_targets.txt \
--outputExcludedSamples $work_dir/xhmm/$pre.filtered_centered.RD.txt.filtered_samples.txt \
--minTargetSize 10 --maxTargetSize 10000 \
--minMeanTargetRD 10 --maxMeanTargetRD 500 \
--minMeanSampleRD 25 --maxMeanSampleRD 200 \
--maxSdSampleRD 150
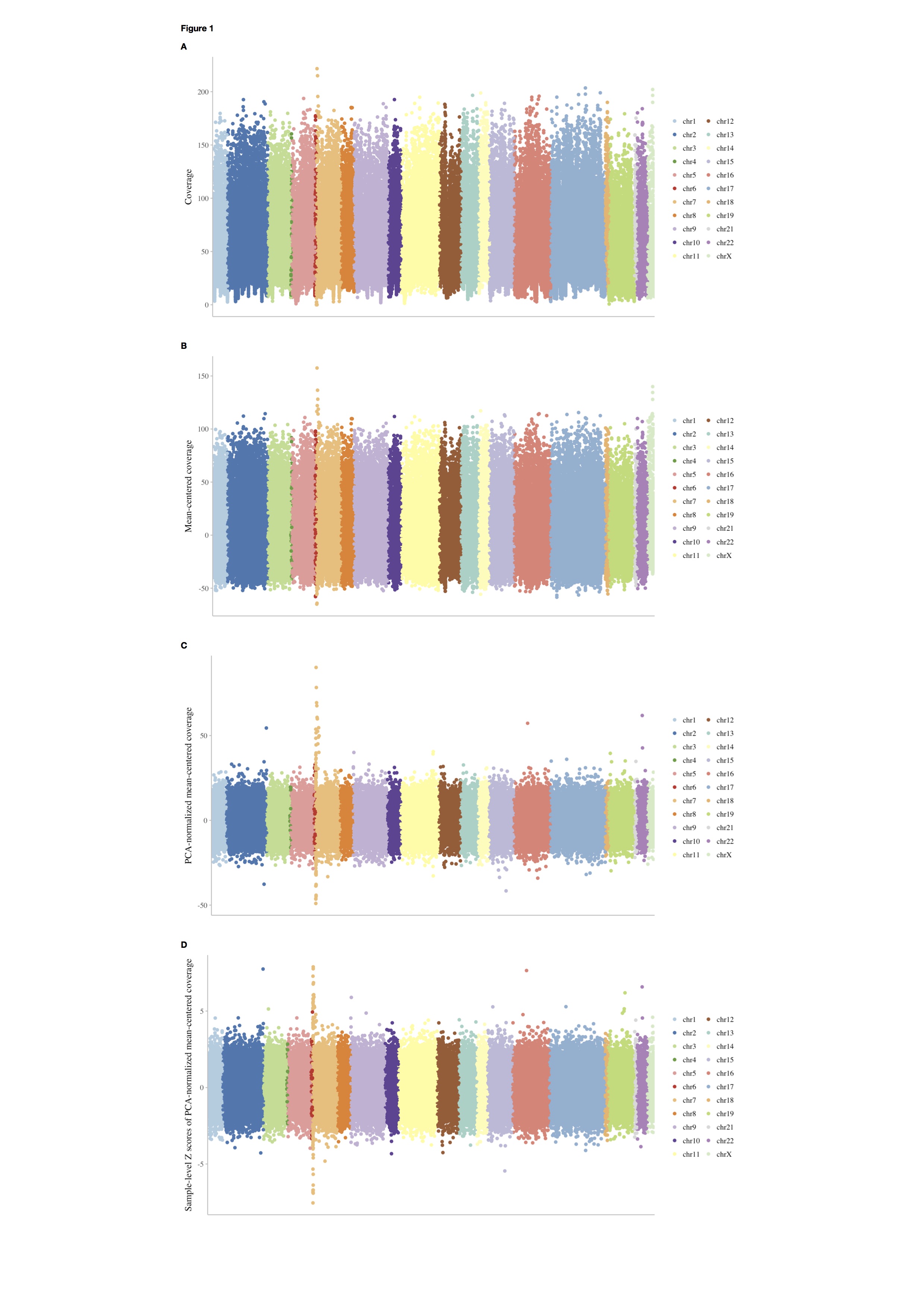
Raw coverage (RD.txt) and the output mean-centered coverage after filtering (filtered_centered.RD.txt) are shown in Figure 1A and 1B.
2.2) Run principal component analysis (PCA) on mean-centered data, and then normalizes the data using PCA information.
PCA analysis is used to determine the strongest independent ways (principal components) in which the data varies and remove the strongest signals that are driven by factors that do not reflect true CNV.
In this case, the first 2 PCs are removed from the datasets.
########
echo "Runs PCA on mean-centered data. Good luck."
#######
$xhmm --PCA -r $work_dir/xhmm/$pre.filtered_centered.RD.txt --PCAfiles $work_dir/xhmm/$pre.RD_PCA
#######
echo "Normalizes mean-centered data using PCA information. Good luck."
#######
$xhmm --normalize -r $work_dir/xhmm/$pre.filtered_centered.RD.txt --PCAfiles $work_dir/xhmm/$pre.RD_PCA \
--normalizeOutput $work_dir/xhmm/$pre.PCA_normalized.txt \
--PCnormalizeMethod PVE_mean --PVE_mean_factor 0.7
In the above codes, the first step outputs 3 files to be used in the next step:
DATA.RD_PCA.PC.txt: the data projected into the principal components
DATA.RD_PCA.PC_LOADINGS.txt: the loadings of the samples on the principal components
DATA.RD_PCA.PC_SD.txt: the variance of the input read depth data in each of the principal components
In the second step, the number of principal components to be removed is determined by the --PCnormalizeMethod PVE_mean --PVE_mean_factor 0.7 argument, which indicates that any principal component in which the data variance is greater than 0.7 times the mean variance over all components will be removed. This argument can be changed to retain additional components or remove more components for normalization.
The coverage after PCA normalization is shown in Figure 1C.
2.3) Z-score normalization and further filtering.
The PCA-normalized coverage data might still have some targets that have very high variance that may be reflective of failed normalization, thus, they are removed in this step. Note, however, that even after this filtering step, exon targets with very high variance in the original read depths still remain with relatively high (though attenuated) variance in depth after normalization, since large differences between samples and targets are by necessity maintained in order to preserve existing CNV signal as well. Then, for each sample, a z-score is calculated by centering relative to all target depths in that sample.
Next, the same targets and samples that are removed during the normalization process are also removed in the original dataset. This matrix will be used for annotation purposes in the subsequent CNV discovery steps.
#######
echo "Filters and z-score centers (by sample) the PCA-normalized data. Good luck."
#######
$xhmm --matrix -r $work_dir/xhmm/$pre.PCA_normalized.txt --centerData --centerType sample --zScoreData \
-o $work_dir/xhmm/$pre.PCA_normalized.filtered.sample_zscores.RD.txt \
--outputExcludedTargets $work_dir/xhmm/$pre.PCA_normalized.filtered.sample_zscores.RD.txt.filtered_targets.txt \
--outputExcludedSamples $work_dir/xhmm/$pre.PCA_normalized.filtered.sample_zscores.RD.txt.filtered_samples.txt \
--maxSdTargetRD 30
#######
echo "Filters original read-depth data to be the same as filtered, normalized data. Good luck."
#######
$xhmm --matrix -r $work_dir/xhmm/$pre.RD.txt \
--excludeTargets $work_dir/xhmm/$pre.filtered_centered.RD.txt.filtered_targets.txt \
--excludeTargets $work_dir/xhmm/$pre.PCA_normalized.filtered.sample_zscores.RD.txt.filtered_targets.txt \
--excludeSamples $work_dir/xhmm/$pre.filtered_centered.RD.txt.filtered_samples.txt \
--excludeSamples $work_dir/xhmm/$pre.PCA_normalized.filtered.sample_zscores.RD.txt.filtered_samples.txt \
-o $work_dir/xhmm/$pre.same_filtered.RD.txt
The coverage after Z score normalization is shown in Figure 1D.
2.4) Genotyping CNVs.
A hidden Markov model (HMM) with certain properties that were derived from a large-scale trio data set is applied in this step. The parameters, which determine the rate and length of the CNV called, are specified in params.txt (1e-8 6 70 -3 1.00 0 1.00 3 1.00)
The 9 values in this file correspond to quantities that respectively define the:
- Exome-wide CNV rate
- Mean number of targets in a CNV call
- Mean distance between targets within a CNV (in KB)
- Mean of DELETION z-score distribution
- Standard deviation of DELETION z-score distribution
- Mean of DIPLOID z-score distribution
- Standard deviation of DIPLOID z-score distribution
- Mean of DUPLICATION z-score distribution
- Standard deviation of DUPLICATION z-score distribution
These parameters are used in the HMM for CNV discovery and genotyping. They may need modification to suit the needs of particular project. For example, to increase the number of CNV calls, increase the first parameter from its default value of 1e-8, e.g., to 1e-7.
The parameters used for the current datasets are (1e-8 6 70 -3 1.1 0 0.9 3 1.1)
#######
echo "Discovers CNVs in normalized data. Good luck."
#######
$xhmm --discover -p $param_adjusted \
-r $work_dir/xhmm/$pre.PCA_normalized.filtered.sample_zscores.RD.txt -R $work_dir/xhmm/$pre.same_filtered.RD.txt \
-c $work_dir/xhmm/$pre.xcnv -a $work_dir/xhmm/$pre.aux_xcnv -s $work_dir/xhmm/$pre
Output file (.xcnv):
SAMPLE CNV INTERVAL KB CHR MID_BP TARGETS NUM_TARG Q_EXACT Q_SOME Q_NON_DIPLOID Q_START Q_STOP MEAN_RD MEAN_ORIG_RD
NA12889_S61 DUP chr7:6017194-6018339 1.15 chr7 6017766 412..413 2 67 88 88 20 25 6.33 135.48
NA18873_S38 DUP chr7:6017194-6018339 1.15 chr7 6017766 412..413 2 50 50 50 6 29 5.46 59.16
HG00451_S34 DUP chr7:6012999-6048725 35.73 chr7 6030862 411..425 15 10 99 99 1 53 4.79 157.75
NA18609_S49 DUP chr7:6017194-6018339 1.15 chr7 6017766 412..413 2 25 36 36 22 7 4.96 107.23
HG00692_S10 DEL chr7:6012999-6022639 9.64 chr7 6017819 411..414 4 7 99 99 23 5 -4.84 16.56
HG01872_S48 DUP chr22:29115357-29121386 6.03 chr22 29118371 1698..1700 3 14 70 70 14 8 4.52 136.17
HG00684_S13 DUP chr7:6012999-6018339 5.34 chr7 6015669 411..413 3 5 99 99 5 21 5.10 79.65
NA18622_S55 DUP chr2:233198528-233208282 9.76 chr2 233203405 212..218 7 30 99 99 14 50 3.72 76.54
NA18558_S39 DEL chr15:80454562-80460700 6.14 chr15 80457631 1162..1164 3 35 39 39 4 27 -4.04 29.29
HG00629_S27 DEL chr7:6012999-6018339 5.34 chr7 6015669 411..413 3 21 99 99 21 5 -5.13 12.96
NA18624_S59 DEL chr7:6012999-6018339 5.34 chr7 6015669 411..413 3 3 99 99 3 7 -4.88 21.31
HG02522_S57 DUP chr7:6012999-6018339 5.34 chr7 6015669 411..413 3 20 41 41 21 13 4.11 67.96
NA11995_S51 DEL chr7:6012999-6018339 5.34 chr7 6015669 411..413 3 9 99 99 9 27 -5.21 0.15
NA07357_S45 DUP chr19:45855446-45873862 18.42 chr19 45864654 1621..1643 23 5 99 99 10 5 3.03 51.15
NA18628_S59 DUP chr7:6012999-6018339 5.34 chr7 6015669 411..413 3 9 37 37 9 8 3.96 38.32
HG00634_S26 DUP chr16:23614761-23615012 0.25 chr16 23614886 1247..1247 1 30 30 30 30 23 7.65 155.58
NA18572_S44 DUP chr9:271606-289600 18.00 chr9 280603 560..562 3 48 80 80 48 15 4.80 105.55
Each line is one CNV called in an individual. Column meaning:
- SAMPLE sample name in which CNV was called
- CNV type of copy number variation (DEL or DUP)
- INTERVAL genomic range of the called CNV
- KB length in kilobases of called CNV
- CHR chromosome name on which CNV falls
- MID_BP the midpoint of the CNV
- TARGETS the range of the target indices over which the CNV is called
- NUM_TARG # of exome targets of the CNV
- Q_EXACT Phred-scaled quality of the exact CNV event along the entire interval
- Q_SOME Phred-scaled quality of some CNV event in the interval
- Q_NON_DIPLOID Phred-scaled quality of not being diploid, i.e., DEL or DUP event in the interval
- Q_START Phred-scaled quality of “left” breakpoint of CNV
- Q_STOP Phred-scaled quality of ‘right” breakpoint of CNV
- MEAN_RD Mean normalized read depth (z-score) over interval
- MEAN_ORIG_RD Mean read depth (# of reads) over interval
3) Compare with ground truth
#######
echo "Get ground truth CNV from 1000G"
#######
CNV_1000G=../1000g/estd219_1000_Genomes_Consortium_Phase_3_Integrated_SV.GRCh37.submitted.variant_call.germline.CNV.txt
sed 's/_.*//' $pre.samplesID.filter > 1000GsampleID.exclude # get the list of samples IDs to be excluded because of coverge issues
sed 's/_.*//' ../sampleID > 1000GsampleID # get the list of all samples IDs
less $pre.targetsID.filter | sed 's/:/\t/;s/-/\t/;s/chr//' |awk 'OFS="\t"{$2=$2-1;print $0}' | sort -k1,1n -k2,2n | subtractBed -a $interval_file -b - > $target.1000G.bed # get the target regions after removing regions with abnormal coverage
Extract.pl 1000GsampleID 1 $CNV_1000G 4 | Exclude.pl 1000GsampleID.exclude 1 - 4 > $pre.1000G.cnv.tmp # get CNVs in selected samples
intersectBed -a $pre.1000G.cnv.tmp -b $target.1000G.bed > $pre.1000G.cnv.tmpa # get CNVs in selected targets
./cnv.adjectmerge.pl $pre.1000G.cnv.tmpa | sort -k1,1n -k2,2n > $pre.1000G.cnv.txt # merge multiple record of the same CNV into one
cat 118bam.105genespure.1000G.cnv.txt
2 233200457 233208282 NA18622 DUP DUP_gs_CNV_2_233200004_233214555_NA18622
7 6031568 6038922 HG00451 DUP DUP_gs_CNV_7_6031005_6039619_HG00451
9 271605 289600 NA18572 DUP DUP_gs_CNV_9_224074_289974_NA18572
15 80454561 80460700 NA18558 DEL UW_VH_20565_NA18558
16 23614760 23615012 HG00634 DUP DUP_gs_CNV_16_23608128_23616407_HG00634
19 45854860 45873862 NA07357 DUP DUP_gs_CNV_19_45845933_45876990_NA07357
19 45854860 45873862 NA07357 DUP DUP_uwash_chr19_45826405_45907191_NA07357
22 29115356 29121386 HG01872 DUP DUP_gs_CNV_22_29114293_29125290_HG01872
Comparing XHMM results with ground truth, we can see that except the one region in chr7 (the region has high-variance in coverage), CNV calls in all other regions are concordant.
ExomeDepth
ExomeDepth runs in three steps.
- Calculate read depth across the datasets, which is similar to XHMM.
- Generate a panel of reference samples that are copy-number neutral.
- For each test sample, compare it against the panel of reference samples, and call CNVs.
Input:
BAM files, interval file, genome fasta file, reference sample IDs (multiple), test sample ID (single)
In the trial, 84 samples that fulfill the following criteria are chosen as reference samples:
- Do not have CNV event in 1000 Genome
- Do not have CNV event in XHMM results
- Sufficient read depth (Mean Sample RD: 25~200)
Scripts:
library(ExomeDepth)
#######
#echo "prepare input bam, bed, and genome fasta files"
#######
bedFile <- file.path("/home/database/targetBed", "105genespure.txt")
targets <- read.table(bedFile,head=T)
b37<-file.path("/home/database/b37","human_g1k_v37_decoy.fasta")
bamName <- list.files("/home/zhengh/projects/CNV/105genes/bam", pattern = '*.bam$')
bamFile <- file.path("/home/zhengh/projects/CNV/105genes/bam",bamName)
#######
#echo "get counts for target regions"
#######
TargetCount <- getBamCounts(bed.frame = targets,bam.files = bamFile,include.chr = FALSE,referenceFasta = b37)
TargetCount.df <- as(TargetCount[, colnames(TargetCount)], 'data.frame')
#######
#echo "prepare reference samples"
#######
sampleID.nocnv<-scan("/home/zhengh/projects/CNV/105genes/xhmm/sampleID.noCNV",quiet=T,what="character")
my.ref.samples <- paste0(sampleID.nocnv,".mapped.dedupped.realigned.recalled.bam")
my.reference.set <- as.matrix(TargetCount.df[, my.ref.samples])
#######
#echo "CNV calling per sample"
#######
sampleID<-scan("/home/zhengh/projects/CNV/105genes/ExomeDepth/samples",quiet=T,what="character")
for (Sample in sampleID){
my.test <- TargetCount.df[,paste0(Sample,".mapped.dedupped.realigned.recalled.bam")]
my.choice <- select.reference.set (test.counts = my.test,
reference.counts = my.reference.set,
bin.length = (TargetCount.df$end - TargetCount.df$start)/1000,
n.bins.reduced = 10000)
my.matrix <- as.matrix( TargetCount.df[, my.choice$reference.choice, drop = FALSE])
my.reference.selected <- apply(X = my.matrix, MAR = 1, FUN = sum)
all.exons <- new('ExomeDepth',
test = my.test,
reference = my.reference.selected,
formula = 'cbind(test, reference) ~ 1')
all.exons <- CallCNVs(x = all.exons,
transition.probability = 10^-4,
chromosome = TargetCount.df$space,
start = TargetCount.df$start,
end = TargetCount.df$end,
name = TargetCount.df$names)
if(nrow(all.exons@CNV.calls)>0){
output.file <- paste0(Sample,".cnv.txt")
write.table(file = file.path("/home/zhengh/projects/CNV/105genes/ExomeDepth/out",output.file),x = cbind(Sample,all.exons@CNV.calls),row.names = FALSE,quote=F,sep="\t")
}
}
Output:
cat *.txt | egrep -v 'Sample' | sort -k8n |cut -f1,4,9
HG02522_S57 duplication chrX:154002844-154004606
NA18622_S55 duplication chr2:233198528-233208282
HG00451_S34 duplication chr6:26092804-26094703
HG00451_S34 duplication chr7:6012999-6048725
HG00629_S27 deletion chr7:6012999-6018339
HG00684_S13 duplication chr7:6017194-6018339
HG00692_S10 deletion chr7:6012999-6018339
HG01872_S48 deletion chr7:6012999-6013213
NA11995_S51 deletion chr7:6012999-6018339
NA12889_S61 duplication chr7:6017194-6018339
NA18609_S49 duplication chr7:6017194-6018339
NA18624_S59 deletion chr7:6012999-6018339
NA18873_S38 duplication chr7:6017194-6018339
NA18572_S44 duplication chr9:214926-289600
HG01872_S48 duplication chr11:118955719-118955888
NA11995_S51 deletion chr15:40493079-40493221
NA18558_S39 deletion chr15:80454562-80460700
HG00634_S26 duplication chr16:23614761-23615012
NA07357_S45 duplication chr19:45855446-45873862
HG01872_S48 duplication chr22:29115357-29121386
Similar to XHMM result, the region in chr7 is quite ambiguous and may not be trustable. Except for chr7 regions, ExomeDepth identifies all the other CNV events in the ground truth 1000 Genome data. Besides, ExomeDepth also detects extra CNVs not present in 1000 Genome and XHMM results, and the accuracy remains to be evaluated.
CODEX
CODEX takes multiple samples as input.
Input:
BAM files, bed file, and sample IDs
Script:
library(CODEX2)
setwd("/home/zhengh/projects/CNV/105genes/CODEX")
######################################################
######################################################
#### ####
#### get coverage, gc, mapp ####
#### ####
######################################################
######################################################
bamName <- list.files("/home/zhengh/projects/CNV/105genes/bam", pattern = '*.bam$')
bamFile <- file.path("/home/zhengh/projects/CNV/105genes/bam",bamName)
sampname <- as.matrix(read.table(file.path("/home/zhengh/projects/CNV/105genes", "sampleID")))
bedFile <- file.path("/home/database/targetBed", "105genes.primary_targets.bed")
projectname<-"CNV_prenetics"
chr=1
# get bam directories, read in bed file, get sample names
bambedObj=getbambed(bamdir=bamFile,
bedFile=bedFile,
sampname=sampname,
projectname=projectname,chr)
bamdir=bambedObj$bamdir; sampname=bambedObj$sampname; ref=bambedObj$ref; projectname=bambedObj$projectname;chr=bambedObj$chr
# get raw depth of coverage
coverageObj=getcoverage(bambedObj,mapqthres=20)
Y=coverageObj$Y; readlength=coverageObj$readlength
# get gc content
gc=getgc(chr,ref)
# get mappability
mapp=getmapp(chr,ref)
ref.all=bambedObj$ref
Y.all=coverageObj$Y
gc.all=gc
mapp.all=mapp
chr.all=rep(chr,length=length(mapp))
targ.chr <- unique(as.matrix(read.table(bedFile, sep = "\t")[,1]))
for(chr in 2:23){
if(chr==23){chr='X'}
if(!is.element(chr,targ.chr)) next
# get bam directories, read in bed file, get sample names
bambedObj=getbambed(bamdir=bamdir,
bedFile=bedFile,
sampname=sampname,
projectname=projectname,chr)
bamdir=bambedObj$bamdir; sampname=bambedObj$sampname; ref=bambedObj$ref; projectname=bambedObj$projectname;chr=bambedObj$chr
# get raw depth of coverage
coverageObj=getcoverage(bambedObj,mapqthres=20)
Y=coverageObj$Y; readlength=coverageObj$readlength
# get gc content
gc=getgc(chr,ref)
# get mappability
mapp=getmapp(chr,ref)
ref.all=c(ref.all,bambedObj$ref)
Y.all=rbind(Y.all,coverageObj$Y)
gc.all=c(gc.all,gc)
mapp.all=c(mapp.all,mapp)
chr.all=c(chr.all,rep(chr,length=length(mapp)))
}
save.image(file=paste(projectname,'_','coverage','.rda',sep=''))
######################################################
######################################################
#### ####
#### normalize ####
#### ####
######################################################
######################################################
load(paste(projectname,'_coverage.rda',sep=''))
ref.all
length(chr.all)
dim(Y.all)
length(gc.all)
length(mapp.all)
gene.all=as.matrix(read.table('/home/database/targetBed/105genes.capture_targets.bed',head=F,sep='\t')[,4])
length(gene.all)
Y=Y.all
ref=ref.all
gc=gc.all
mapp=mapp.all
gene=gene.all
cov_thres1 = 20
cov_thres2 = 500
sample_median_thres = 25
qcObj=qc(Y,sampname,chr,ref,mapp,gc,cov_thresh=c(cov_thres1,cov_thres2),length_thresh=c(20,2000),mapp_thresh=0.9,gc_thresh=c(20,80))
Y_qc=qcObj$Y_qc; sampname_qc=qcObj$sampname_qc; gc_qc=qcObj$gc_qc; mapp_qc=qcObj$mapp_qc; ref_qc=qcObj$ref_qc ; qcmat=qcObj$qcmat
dim(Y_qc)
length(gc_qc)
length(mapp_qc)
gene_qc=gene[which(as.logical(qcmat[,4])==TRUE)]
chr_qc=chr.all[which(as.logical(qcmat[,4])==TRUE)]
length(gene_qc)
length(sampname_qc)
sampfilter=apply(Y_qc,2,median)>=sample_median_thres
sampname_qc=sampname_qc[sampfilter]
Y_qc=Y_qc[,sampfilter]
rm(qcObj)
normObj <- normalize(Y_qc, gc_qc, K = 1:10)
Yhat <- normObj$Yhat; AIC <- normObj$AIC; BIC <- normObj$BIC ;RSS <- normObj$RSS; K <- normObj$K
save.image(file=paste(projectname,'_','normalize','.rda',sep=''))
######################################################
######################################################
#### ####
#### segment ####
#### ####
######################################################
######################################################
load(paste(projectname,'_normalize.rda',sep=''))
choiceofK(AIC,BIC,RSS,K,filename=paste(projectname,'_choiceofK','.pdf',sep=''))
dim(Y_qc)
length(ref_qc)
length(sampname_qc)
dim(Yhat[[2]])
length(chr_qc)
#plot(K, RSS, type = "b", xlab = "Number of latent variables")
#plot(K, AIC, type = "b", xlab = "Number of latent variables")
#plot(K, BIC, type = "b", xlab = "Number of latent variables")
optK=which.max(BIC)
optK=1
source('/home/zhengh/projects/CNV/105genes/CODEX/segment_targeted.R')
finalcall=matrix(ncol=14)
colnames(finalcall)=c('sample_name','chr','gene','cnv',
'st_bp','ed_bp','length_kb',
'st_exon','ed_exon','raw_cov',
'norm_cov','copy_no','lratio',
'mBIC')
for(genei in unique(gene_qc)){
cat('Segmenting gene',genei,'\n')
geneindex=which(gene_qc==genei)
yi=Y_qc[geneindex,]
yhati=Yhat[[optK]][geneindex,]
refi=ref_qc[geneindex]
chri=chr_qc[geneindex][1]
finalcalli=segment_targeted(yi, yhati, sampname_qc, refi, genei, chri, lmax=length(geneindex), mode='fraction')
finalcall=rbind(finalcall,finalcalli)
}
finalcall=finalcall[-1,]
cn=(as.numeric(as.matrix(finalcall[,'copy_no'])))
cn.filter=(cn<=1.7)|(cn>=2.3)
finalcall=finalcall[cn.filter,]
length_exon=as.numeric(finalcall[,'ed_exon'])-as.numeric(finalcall[,'st_exon'])+1
finalcall=cbind(finalcall[,1:7],length_exon,finalcall[,10:14])
write.table(finalcall, file = paste( projectname,'_', optK, '_CODEX_frac.txt',
sep=''), sep='\t', quote=F, row.names=F)
save.image(file=paste(projectname,'_','finalcall','.rda',sep=''))
Output:
There are two many CNV calls and false discovery rate is very high.
Read pair-based methods
Several methods based on read pairs, including BreakDancer, Pindel, and BreakSeq2 have been utilized to examine the datasets. As expected, these methods don’t perform well for targeted sequencing datasets. Due to that fact that most breakpoints do not fall into exon regions, these methods can hardly identify true CNVs, and the results are mainly false positives. Therefore, the detailed scripts and results are not shown here. They can be found in /home/zhengh/projects/CNV/105genes/.
Other methods
Besides XHMM, ExomeDepth, CODEX, and several read-pair based methods, two other methods were also tested, but with no meaningful outputs.
- CNVnator. This tool is only suitable for whole genome sequencing.
- CoNIFER. This tool is only suitable for whole exome sequencing or genome sequencing. Targeted sequencing has too few targets.
Conclusion
To identify CNVs from targeted sequencing data, RD-based methods perform better than read pair-based methods. Among the RD-based methods, XHMM and ExomeDepth are both good at CNV detection utilizing a population of samples.
- XHMM works with multiple samples in each stage of the workflow.
- ExomeDepth also need multiple samples at the first stage to build a panel of reference. After the reference panel is ready, ExomeDepth can take in single sample each time, compare it with reference panel, and output CNV calls in the single sample.
- The results of XHMM and ExomeDepth are highly consistent and they both can identify CNVs in 1000 Genome ground truth dataset, except for a few problematic regions. In future workflow, these two methods can be combined to get a more confident CNV callset.
Several aspects need to be paid attention to:
- Sufficient sample size and number of targeted region are needed to accurately model the read depths and detect CNVs.
- The RD-based CNV methods, including XHMM and ExomeDepth , depend on a set of normal samples (without CNV changes) in the datasets to serve as baseline for CNV calling. For example, if all the samples in the datasets have deletions in a specific region, these methods will classify it as systematic problem (failure to capture, mapping problem etc.) and will not output the region as deletion event.
- Fine tuning of the parameters/choices of samples is needed in future workflow to achieve high accuracy.